
Christian Perspectives on Science and Technology, New Series, Vol. 1 (2022), 150–174
Abstract: The lines of biological evolution are documented in the genomic “texts” of species. Phylogenies of texts, both genetic and literary, can be studied by the same methodologies. In each case, scholars use the presence of variants to elucidate the history of their chosen text—whether it be genetic (the four chemical letters inscribed in DNA) or alphabetic (the letters of biblical languages such as Hebrew and Greek). Several conclusions arise. First, genetic and textual variants constitute the data from which phylogenetic trees of organisms and manuscripts (respectively) may be constructed. Second, such analyses assume the existence of (now extinct) ancestral genomes and ancestral texts, providing evidence that such urtexts existed and enable their reconstruction. Third, biological evolution belongs to the category of history, and like all histories, can be understood as development within the created order. Fourth, biological evolution raises questions about divine providence that are similar to questions that arise from any other history. Fifth, theologians need to develop a theology of evolutionary history in the same way as they seek to understand God’s [151] action in biblical history (allowing that only the latter involves personal creatures).
We can often recognise family likenesses. Shared gene variants underlie such resemblances—although shared environments also contribute to similar phenotypes. Genomic data, especially from DNA sequencing, are routinely used to assess the relatedness of individuals and, increasingly, to assess the relatedness of species and the routes by which a single progenitor species can diversify into multiple descendant species.
The phylogenetic development of organisms has been depicted in evolutionary trees for many years. In the genomic era, such trees are constructed using genetic variants. Such powerful genomic markers include chromosome rearrangements and genetic parasites (endogenous retroviruses and transposable elements). These markers outline the transformations of genomes including the route of humanity’s evolutionary past.
But some Christians may find phylogenetic trees disconcerting. For the Bible says we are created, and some believers think that a continuous link with monkey forebears contradicts this claim. Phylogenetic trees indicate that diverse living forms are derived from common ancestors by what appear to be mechanistically describable biological processes. Continuous process seems to rule out direct action of a creator God. Miraculous acts of creation become redundant.
This paper will describe how biological evolution (including ours) is demonstrated by comparing variations in genomes of multiple species. The genomic approach will be expounded by comparing it with a topic familiar to Christians—the application of textual criticism (where criticism means analysis) to describe the history of ancient biblical manuscripts. There is continuity between the genetic texts (genomes) of species as there is between the written texts of manuscript traditions. The evolution of genomes reveals biological history in the [152] same way as the evolution of variants of biblical manuscripts reveals the literary history of those texts. Such progressive transformations are equally histories, developments within God’s created world. Biological evolution and the biblical concept of creation belong to alternative ontological categories, but they are not opposed to each other. While evolution is a freely operating historical process, it is an ever-dependent one, ordained by God.
Chromosome History: Cutting and Pasting
When cells divide, their DNA and associated proteins are packaged in chromosomes, bodies that can be seen using conventional light microscopes. Using appropriate stains, individual chromosomes, and parts thereof, can be identified. The normal human genome consists of forty-six chromosomes including twenty-two pairs of autosomes and two sex chromosomes (XX in females; XY in males).[2]
Closely related species have similarly structured chromosome sets (or karyotypes). By aligning chromosome sets from different species, cytogeneticists can detect differences between them, such as fissions (a chromosome present in one species has split to form two chromosomes in other species), fusions (two chromosomes have joined end-to-end) and reciprocal translocations (chromosomes have exchanged lengths of material). More subtle, but more frequent, rearrangements include inversions (a block of chromosomal material flips 180° with respect to its surrounds), duplications, insertions, and deletions. Stepwise chromosomal rearrangements are familiar natural phenomena, and frequent in cancers.
The alignment of karyotypes from related species demonstrates how one karyotype can be transformed into another by cutting-and-pasting chromosomal material. In addition, such comparative studies allow reconstruction of the karyotypes of common ancestors (now extinct). We could say that we may infer the genomic [153] urtext, the ancestral version from which all derivative extant genome arrangements are derived. In biblical textual criticism, the urtext is the original form of a composition. We could call the ancestral karyotype of a group of species its urkaryotype.
The lines of primate chromosome evolution have been delineated by this microscopic level comparative cytogenetics approach.[3] More remotely, the ancestral eutherian[4] karyotype has been reconstructed, and can be transformed into the ancestral primate karyotype by three fissions and two fusions. The latter can be transformed into the ancestral anthropoid (simian; monkey-ape) karyotype by a fission, a fusion, and a translocation, and from thence into the ancestral hominoid (ape, including human) karyotype by another fission. Morphological comparisons indicate that the hominoid common ancestor had 48 chromosomes. Humans have 46 chromosomes because two chromosomes retained in the chimpanzee genome fused to form human chromosome 2 (via an inversion in each of the two precursor chromosomes that occurred in a human-chimpanzee ancestor).
These insights, obtained by observing chromosomes microscopically, are of relatively low resolution. The era of high-throughput genome sequencing (with computational analysis of sequences) now allows chromosome rearrangements to be identified and mapped at high resolution. Genome evolution can be studied at the level of the genetic text.
The karyotype of the eutherian ancestor, that lived at least 80 million years ago,[5] can be rearranged into the human karyotype by 162 DNA breakage events (of which the most common generated inversions).[6] [154] The karyotypes of long-extinct ancestors intermediate between the eutherian and human have also been deduced from the chromosome complements of extant species. Fig. 1 depicts the number of inferred DNA breakages that converted the karyotype of a eutherian ancestor into the human one.
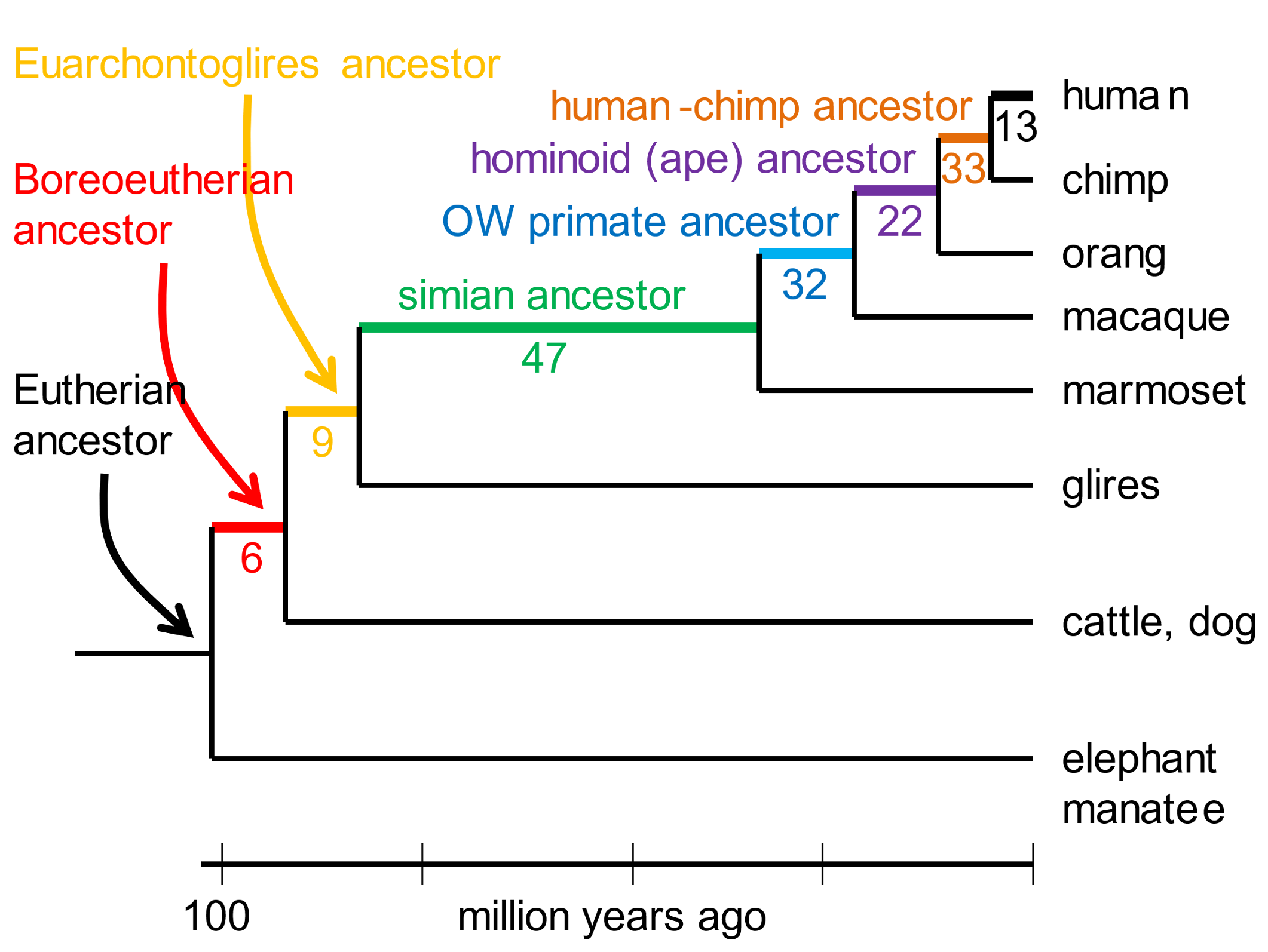
Fig. 1. DNA breakpoints reconstructed from comparing karyotypes of extant species The resolution of analysis was DNA segments ≥300,000 bases long. Glires include rodents and rabbits. Boreoeutherians are a major category of mammals, that include primates, rodents, hoofed mammals and whales, carnivores, and bats. Adapted from Kim et al. (2017), ref. 6.
More recently, genomes of species yet more distant from ours have been sequenced. Alignments including the platypus and echidna genomes (along with karyotypes of marsupials, chicken, and a lizard) have enabled geneticists to reconstruct the karyotype of the mammalian [155] ancestor. Some 165 rearrangements were needed to convert the ancestral mammalian genome, the mammalian genetic urtext, into the human one.[7]
Similar reconstructions based on the karyotypes of extant birds, have indicated the likely karyotypes of a bird ancestor, of a bird-turtle (archelosaur) ancestor, and of a bird-reptile (diapsid) ancestor. As with the mammal karyotype, the chromosome sets are interconvertable by the chromosome rearrangements familiar to geneticists. Seven fissions are needed to convert the turtle karyotype into the avian one.[8]
Karyotypes of birds, of turtles, and of squamates (snakes, lizards) include tiny microchromosomes as well as conventional macrochromosomes. Certain microchromosomes in different species contain the same genes and must have been inherited intact from the same ancestor. They are related also to the microchromosomes which comprise the genome of amphioxus, a little fish-like creature that is classified near the base of the chordate family tree.[9] In birds and reptiles, the number of microchromosomes tends to decrease due to fusions that generate macrochromosomes.[10] Platypuses have several small chromosomes that are products of micro-micro fusions. All other mammals lack microchromosomes, and segments derived from microchromosomes have been fragmented beyond recognition by genome rearrangements.
[156] An ever-increasing database of sequenced genomes, coupled with sophisticated algorithms, is facilitating the reconstruction (at higher resolution) of even more distant ancestral karyotypes. The number of reconstructed ancestral genomes approximates the number of sequenced extant genomes.[11] An example, shown in Fig. 2, depicts how a reconstructed genomic arrangement belonging to an ape ancestor can be rearranged to form the human and (more fragmented) gibbon karyotypes.[12]
An urkaryotype of an ancestor of all multicellular animals may be derived by comparative analysis of more simple animals. Organisms involved in this work included sponges, cnidarians (sea anemones, corals, jellyfish) and bilaterians (worms, molluscs, and chordates).[13]
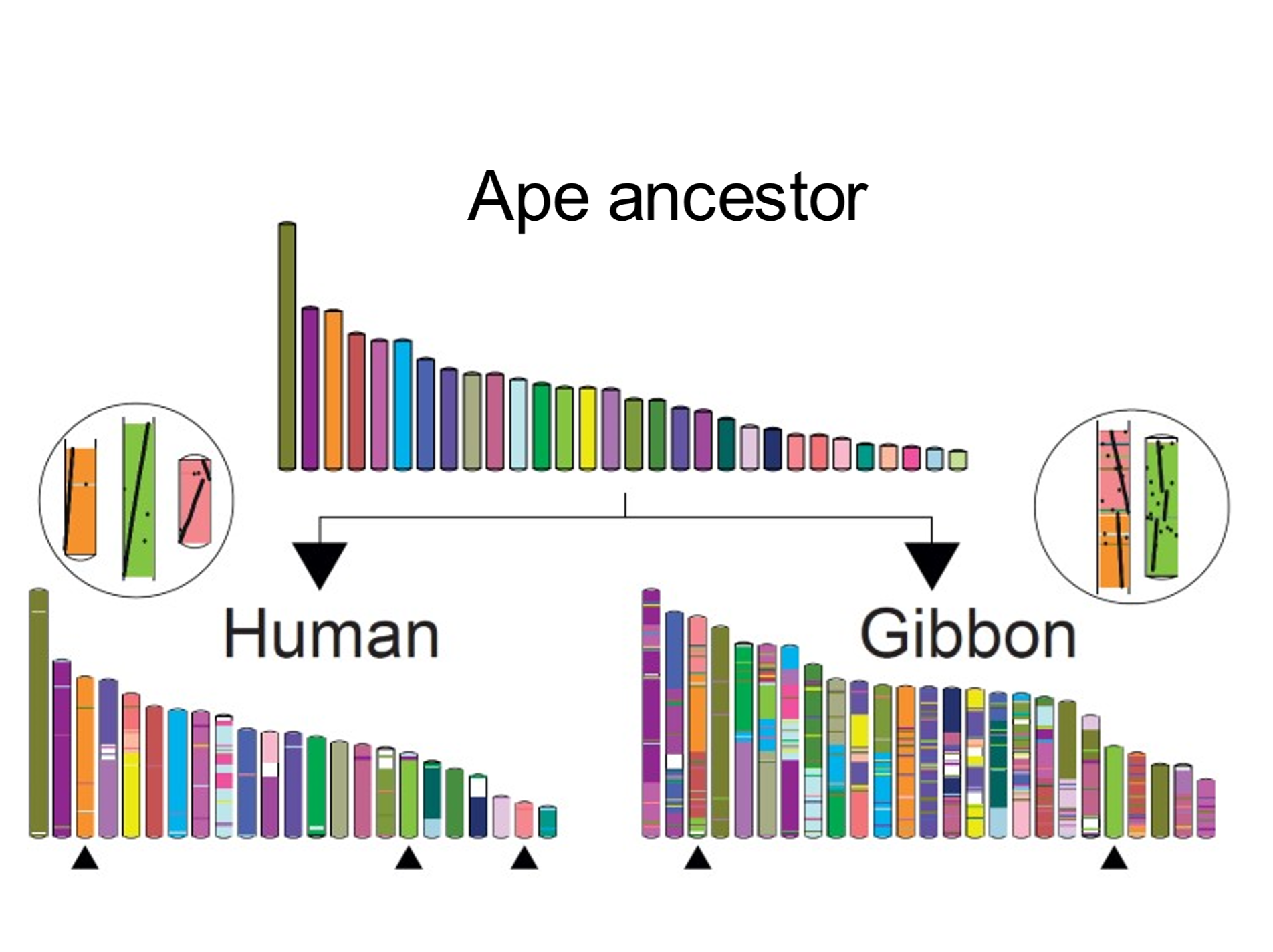
Fig. 2. Cutting-and-pasting chromosomes: converting the ancestral ape karyotype into the human and gibbon karyotypes. The inferred ancestral ape (hominoid) karyotypic arrangement is shown. Each coloured bar represents a block of chromosome material (a contiguous ancestral region). Chromosomal blocks are colour-coded to indicate relation to the ancestral regions. The human karyotype is less rearranged than that of gibbon. From Muffato et al. (2022), ref. 11. Used with kind permission of Dr Roest Crollius.
DNA is chemically simple but informationally rich—a text of extraordinarily dense content. It contains a detailed record of a species’ history. Alignments of the genetic texts belonging to multiple related species or groups of species reveal when novelties appeared. When increasingly remotely related species are used in the intertextual comparisons, a series of ancestral karyotypes can be inferred. [157]
Parasites in Our Genome
Genomes are not static assemblages of genes. New genetic material is constantly added. One source of novel DNA is a category of infectious agents called retroviruses. When a cell is infected, the retroviral genome is copied from RNA into DNA by a retrovirus-encoded enzyme called a reverse transcriptase. Another viral enzyme, an endonuclease, selects (at random) a point in the host DNA (the target site), and splices the retroviral DNA into the host DNA at this site. During this process, the target site is duplicated so as to bracket the new segment of retroviral DNA. If the infected cell is a reproductive cell that can transmit its DNA to future generations, the inserted retroviral DNA may become a feature of the genome of the species. It is said to be endogenous. This process is depicted in Fig. 3 (left scheme).
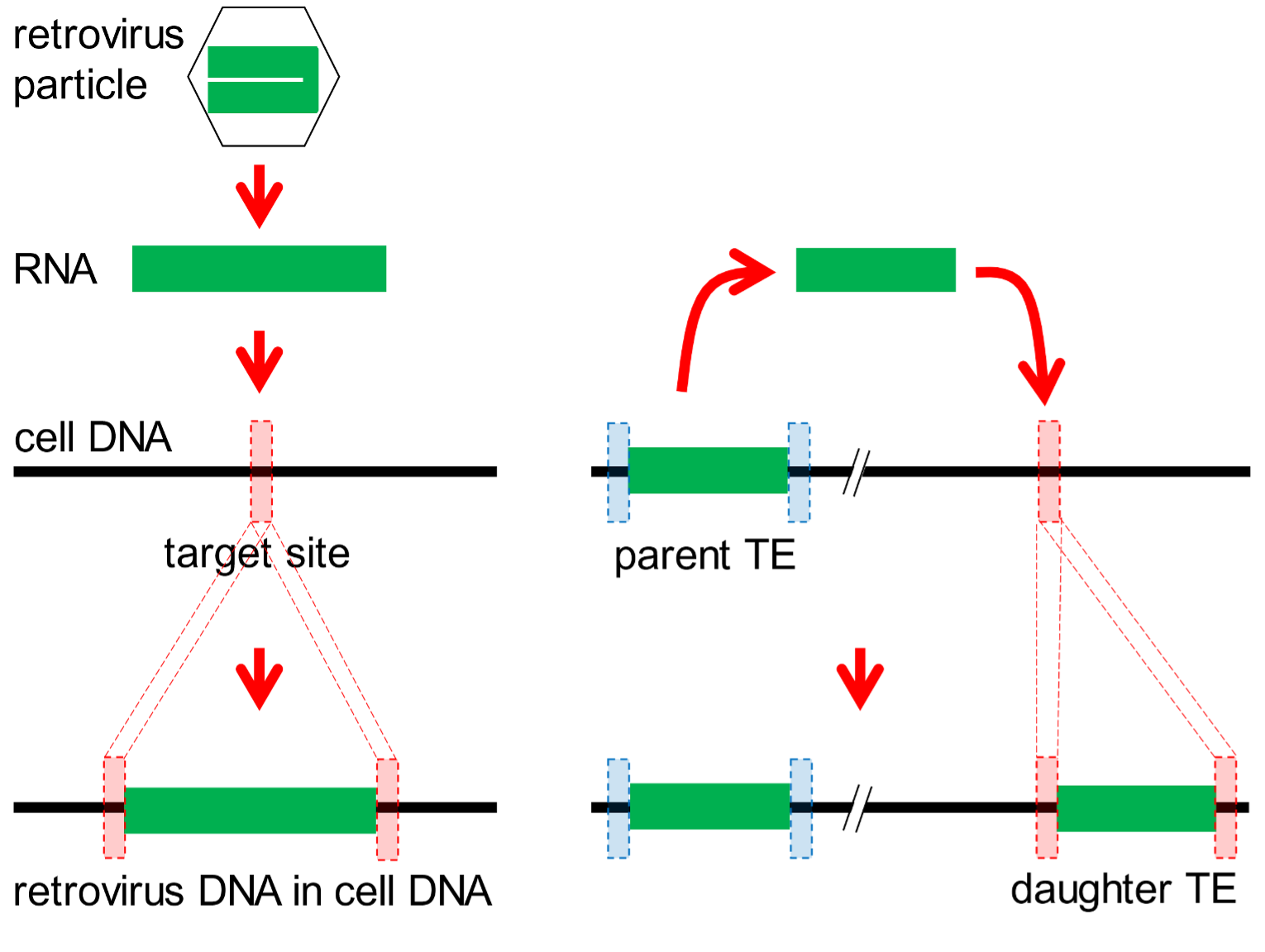
Fig. 3. Agents that modify genomes. Most of our genome is comprised of parasitic units of DNA. These are of two main types. Left: Retroviruses (hexagon) colonize DNA by introducing their RNA (green bar) into chromosomal DNA of infected cells. Viral enzymes select a target site (red dotted box) in the cell DNA at which the retroviral genome is copied into DNA and inserted into the cell’s DNA. During this process, the target site is duplicated. Right: Transposable elements (TEs) are of many types, but typically reproduce in genomes when a parent TE is copied into RNA. Enzymes produced by TEs select a target site elsewhere in the genome (red dotted box) at which the TE RNA is copied into DNA and inserted into the cell’s DNA. Again, the target site is duplicated.
[158] Another broad category of parasitic DNA resides in and colonises genomes. There are hundreds of different subtypes of such parasitic DNAs in genomes such as ours, and collectively they are known as transposable elements (TEs). Most of these multiply by a copy-and-paste strategy using TE-encoded reverse transcriptase/endonuclease enzymes. They also generate target site duplications as they replicate (Fig. 3, right scheme). As with retroviruses, if a new TE is generated in a reproductive [159] cell, it may be passed on to future generations and become a characteristic feature of a species’ genome.
Endogenous retroviruses and TEs together comprise more than fifty percent of our entire genome. The vast majority of these are shared by all human beings. The question arises as to when they entered our genomes. As with studies on chromosome number and structure (karyotype), we can best address this question by aligning the genome sequences of human and other species and ascertaining whether a particular element is shared by multiple species. Such a genomic comparison is exemplified for a TE called an SVA element that is located near the SHPK gene (Fig. 4).[14] The sequence of the four letters (A, C, G, T) that comprise the genetic “text” around the insertion site is presented.
This alignment shows that the SVA element entered primate DNA in an ancestor of the African great apes (human, chimp, bonobo, gorilla). The undisturbed target site is present in Asian apes (orangutan, gibbon), Old World monkeys (four species shown), and New World monkeys (four species).
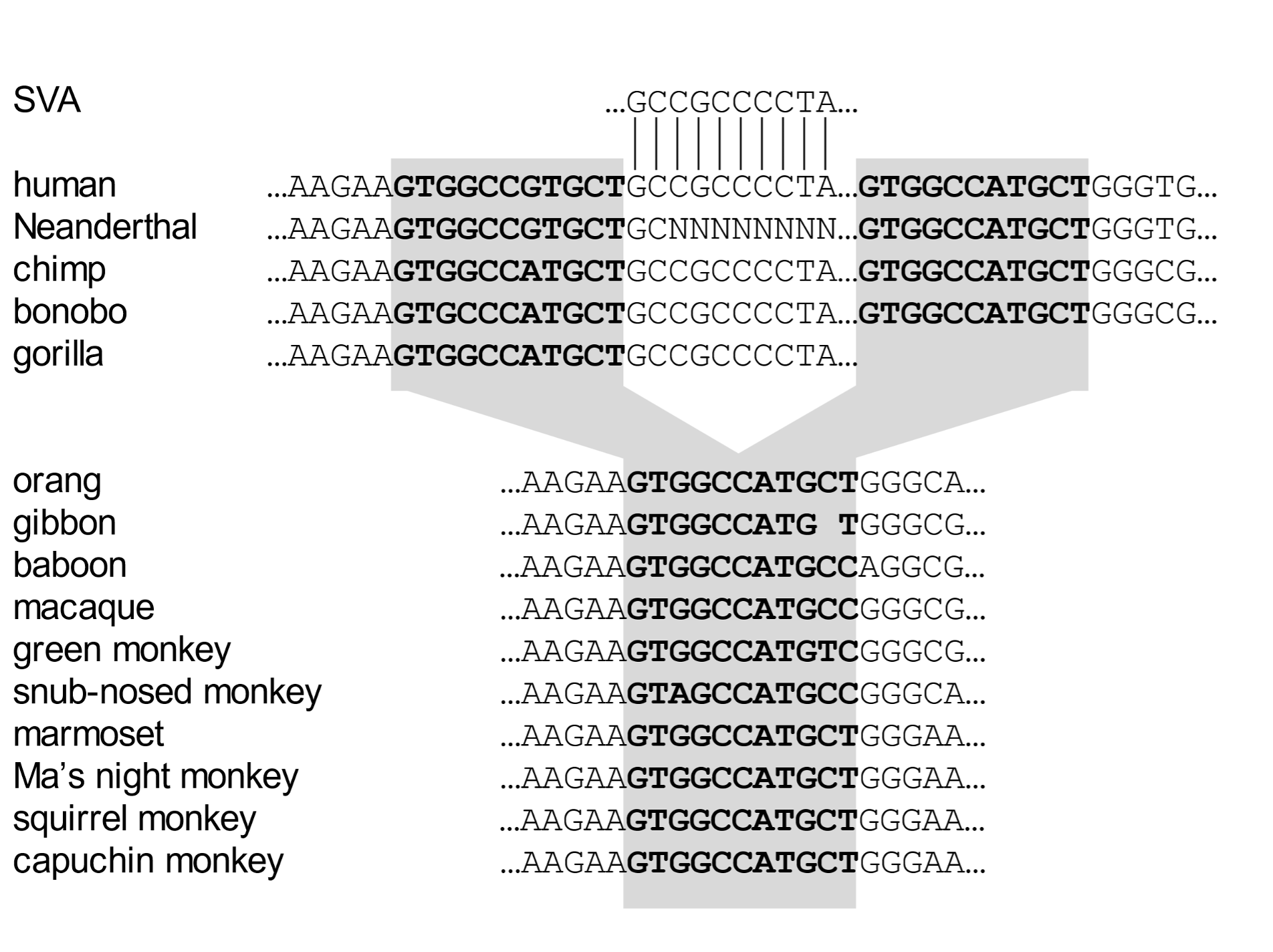
Fig. 4. The insertion site of an SVA element in humans and other primate species The DNA sequence alignment represents a comparison of genetic text that indicates when in primate history the insertion mutation occurred. The SVA element (starting GCCGCCCCTA…) is present between duplicates of the target site (in bold text and shaded) in humans, chimps, bonobos, and gorillas. (In the case of gorillas, right-hand sequences have been deleted). Other species retain the original undisturbed target site. In the human and Neanderthal left-hand target site duplicate, the seventh based has mutated to a G, whereas the same position in all other cases is A. A sequence gap is indicated by “N.” This insert is near the SHPK (sedoheptulokinase) gene. From Price et al. (2021), ref. 14.
SVA elements appeared only in great apes. They were cobbled together from bits of genetic flotsam. They are mutagenic (alter DNA sequences) and certain insertions (depending on their genomic location) cause genetic diseases. Thousands of SVA elements are present in great ape genomes, and the host species of each element is known.[15] Five hundred of these have been selected to create a phylogenetic tree of the great apes (Fig. 5). Most SVA elements have arisen relatively recently and are found only in one species. For example, 98 are found only in the human genome. Forty-four inserts are shared by human and chimp genomes. They were added to the genomes of human-chimp ancestors. [160] Ninety are shared by human, chimp, and gorilla genomes. Each one of these is a powerful demonstration of African great ape monophylicity (descent from the same ancestral linage). And a handful are common to all great ape genomes (but were not present in the selection of inserts depicted in Fig. 5).
Somewhat mysteriously, five SVA inserts are present in human and gorilla genomes, but not that of chimpanzees (Fig. 5, “HG, 5”). At first glance, this is not compatible with the phylogenetic tree. We can account [161] for anomalous insertions by a phenomenon called incomplete lineage sorting. Some anomalous inserts are inevitable, as illustrated in Fig. 5.
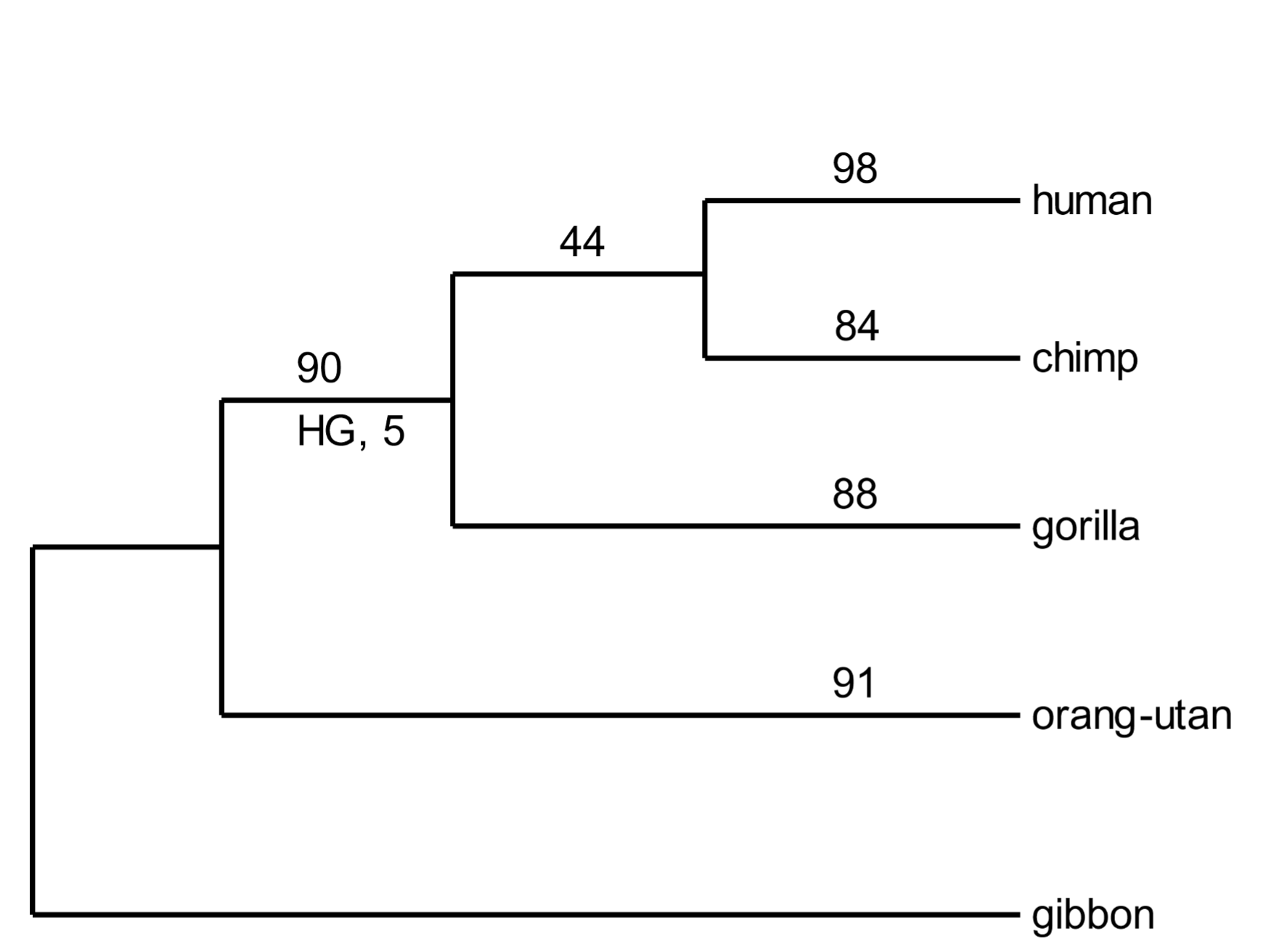
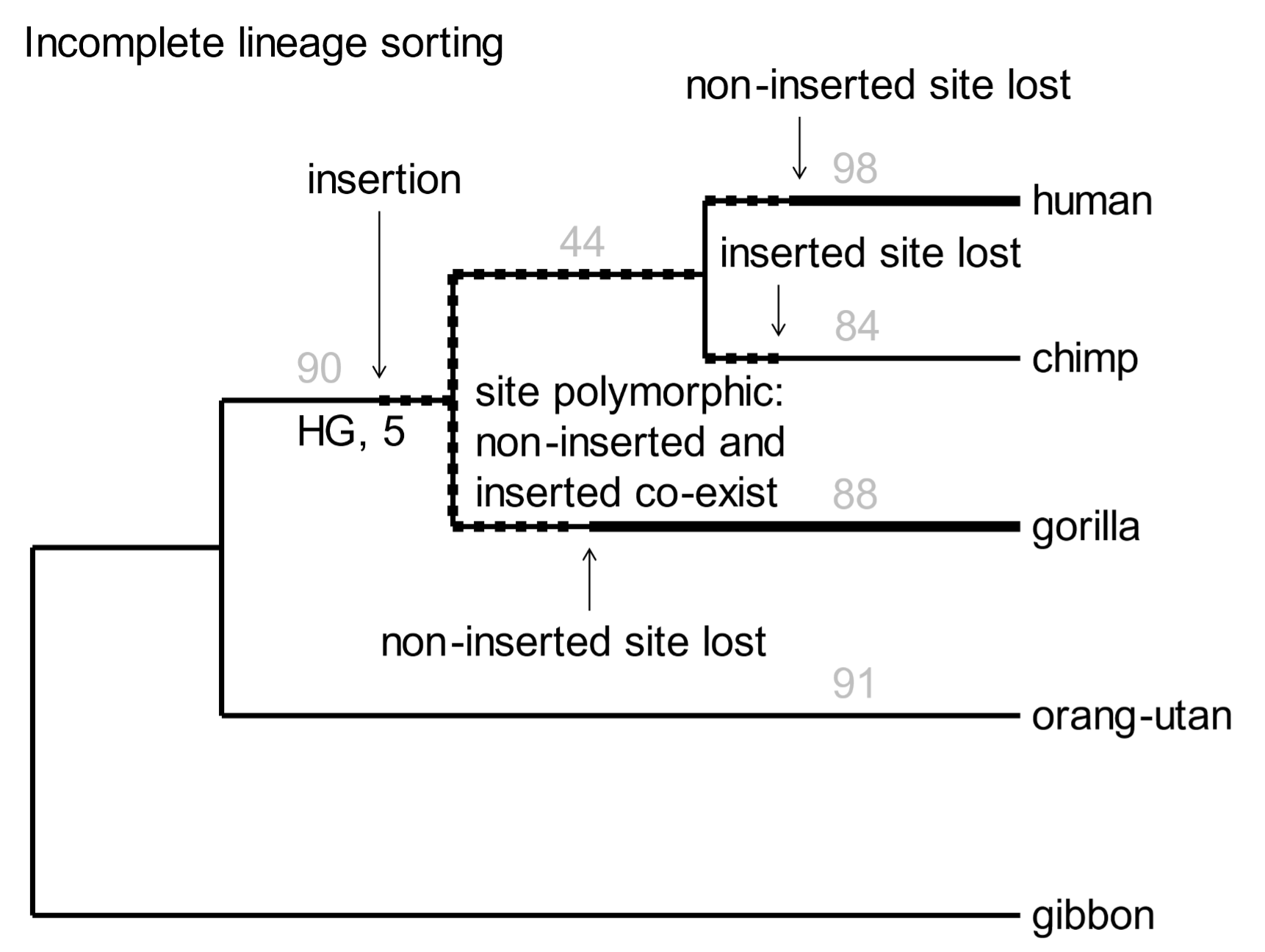
Fig. 5. A phylogenetic tree of great apes based on SVA element insertions Above: In the random selection of SVA elements used above, none was present in humans, chimps, gorillas, and orangutans. However, several have been documented, establishing great ape monophylicity. From Levy et al. (2017), ref. 15, Data fileS1, SupData3 (orthologues table), with 7488 individual SVA insertions. Below: Incomplete lineage sorting occurs when a retrovirus or TE is inserted into germline DNA near the time when species diverge. The original chromosome without the insert, and the derived chromosome with it, will coexist in the population. The site is polymorphic with respect to the inserted element. As the nascent species develop, the chromosome with the insert may randomly drift in frequency so that it may be either lost (only the pre-insertion chromosome is retained) or fixed (the pre-insertion chromosome is lost).
[162] Every event involving the insertion of a transposable element is unique. The new element may be transmitted to members of the family, then to members of the tribe, and eventually to individuals in the wider population. For a considerable period, both the original, pre-inserted sequence and the derived SVA element-containing sequence will be present in the genetic pool of the population. The site is said to be polymorphic—it has two alternative sequence features. If two or more species diverge at this point, the element will be inherited in a polymorphic state in all nascent species. As a result of random drift, it will eventually either be lost, or it will displace the original pre-inserted sequence.[16] In the latter case the new element becomes fixed as a property of the genome. The five SVA inserts noted in Fig. 5 arose in the genomes of human-chimp-gorilla ancestors, were polymorphic when the species diverged, were lost during subsequent chimp history, but retained and fixed in the human and gorilla lineages.
TEs have featured in this discussion because they have had a huge effect on genome evolution and are powerful markers of phylogenetic relationships. The same conclusions are reached by comparative studies using other categories of mutations. For example, simple insertions and deletions in genomes of multiple species corroborate the accepted outline of primate evolution. These uniquely arising mutations demonstrate the monophylicity of the African great apes, and the fact that humans are more closely related to chimps than they are to gorillas.[17]
Incomplete lineage sorting is rampant during bursts of speciation. A mammalian superorder called Euarchontoglires includes five orders: primates, flying lemurs, tree shrews, rodents, and rabbits. Selection of a subset of transposable elements that proliferated [163] during early Euarchontoglires history shows complex patterns of element presence and absence, and of phylogenetic relationships.[18] Similarly, Laurasiatherian mammals include moles, cattle and whales, carnivores, pangolins, horses and bats. Incomplete lineage sorting was rife in the early days of their evolution. A group of informative transposable elements indicates that these orders diverged from the Laurasiatherian ancestral species in complex ways.[19]
A standard phylogenetic tree (such as those of Figs 1 and 5) cannot depict the network of relationships entailed in widespread incomplete lineage sorting. Mathematicians have devised computational methods that can express the complexities of anomalous trees. An informative analysis is that of the SplitsTrees algorithm, which presents the early phylogenetic relationships of diverging species as networks (Fig. 6). These show (for example) that primates are most closely related to flying lemurs, followed by tree shrews; and that carnivores are most closely related to pangolins and then to cattle and whales. [164]
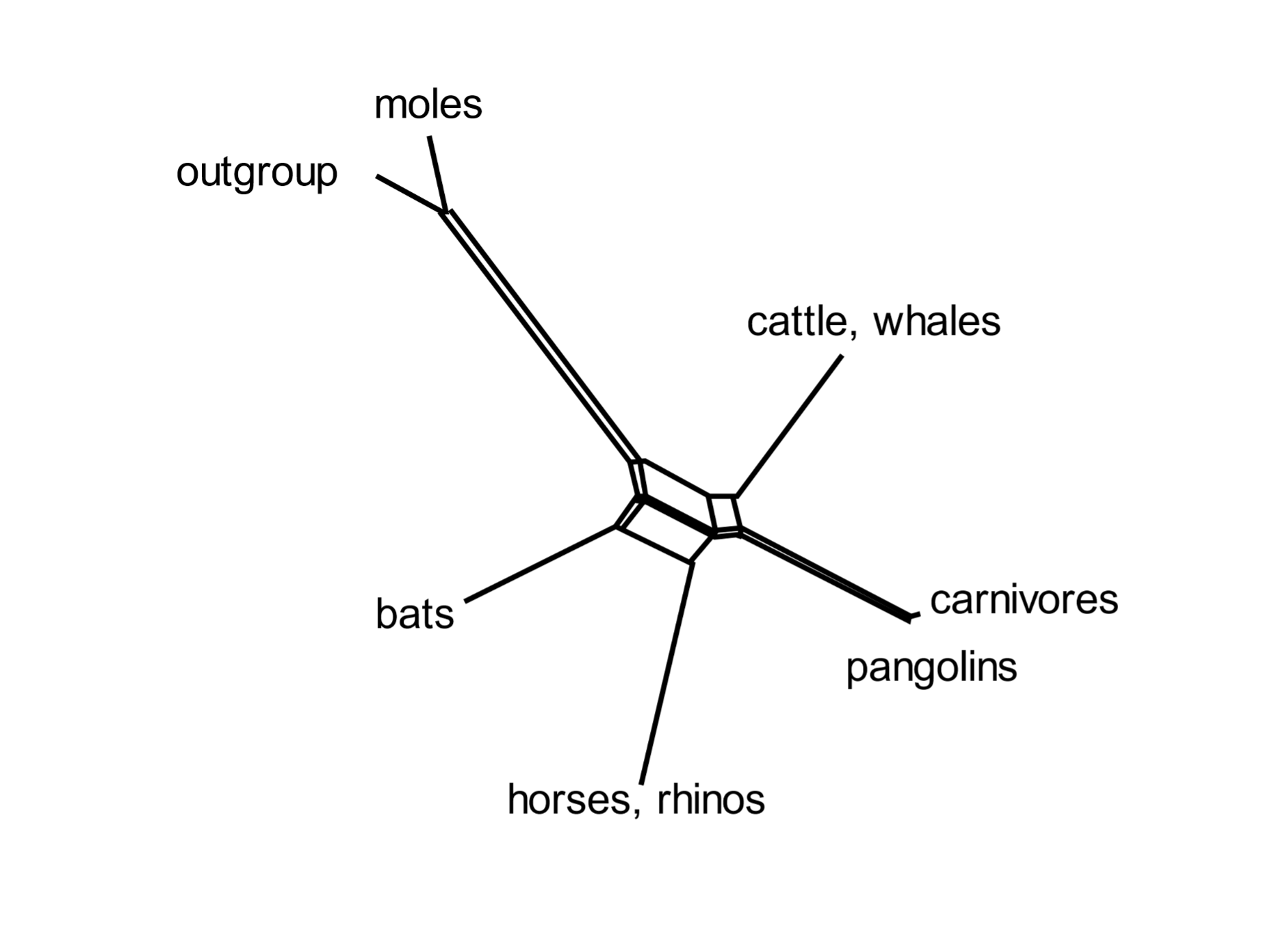
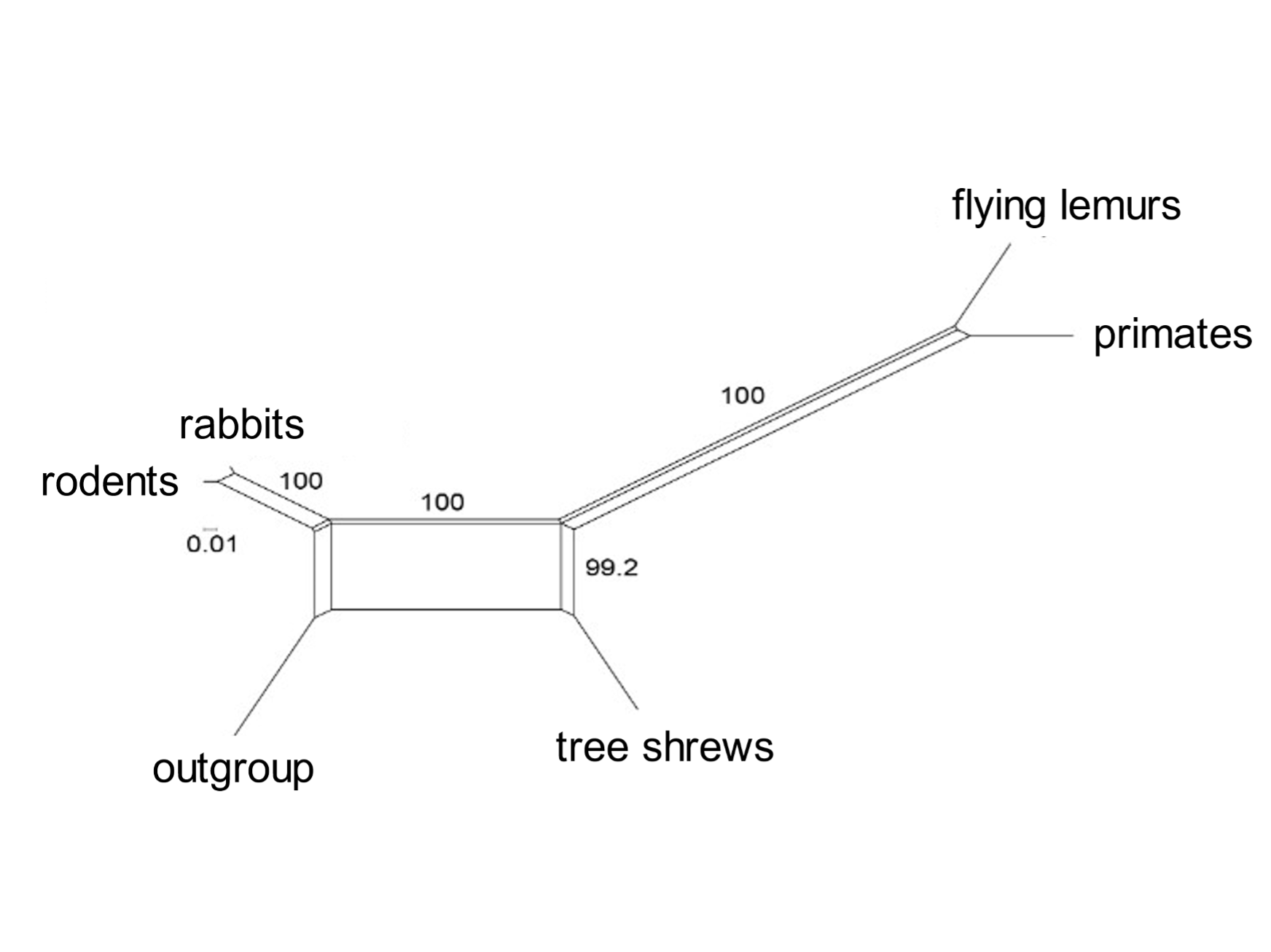
Fig. 6. Incomplete lineage sorting during mammalian evolution: SplitsTrees representation The SplitsTree diagram shows the relationships of the orders included in Euarchontoglires (above) and six orders in Laurasiatheria. It is based on the presence or absence of a set of transposable elements. Both groups show a network of connections during the early burst of speciation. From Doronina, Reising et al. (2022), ref. 18, and Doronina, Hughes et al. (2022), ref. 19.
Phylogenetic Trees Reveal the Shape of History: Comparison with Ancient Manuscripts
The era of comparative genomics has facilitated the application of genetic “textual criticism”—the analysis of changes in the DNA “text” that have occurred during evolution. Biblical textual critics have been doing the same thing with ancient manuscripts for many years. In 1832, Lachmann proposed that manuscripts that share common errors (especially highly distinctive ones) have a common ancestry. This is because the presence of an “indicative error” in two or more manuscripts [165] cannot have been made on two separate occasions.[20] This information may be used to construct family trees (stemmata) of manuscripts.[21] Variants in extant manuscripts can reveal when those novelties arose in (now lost) ancestral manuscripts. These concepts are expanded below.
The genome undergoes several mutations every time a cell replicates its DNA in preparation for cell division. Ancient manuscripts generated by manual copyists undergo textual changes every time a manuscript is copied. In each case, new variants may be preserved in succeeding iterations of the copying process. Genomic texts and ancient written biblical texts thus accumulate mutations or variants with successive copying. Such variants act as markers that can be used to trace the history of the text. The phylogenetics of species and of ancient texts are closely analogous processes.[22] Indeed, the New Testament scholars Wright and Bird speak of “the living process of textual transmission.”[23] Textual difference could be understood “as a stage in a living text’s adaptation to its environment.”[24]
The approach of constructing phylogenetic trees or stemmata using genomes or literary texts requires that the researcher aligns texts from various sources, and notes variants present in them. Some variants will be peculiar to a single text, and others may be represented in multiple different texts, suggesting that those many texts are related.
Genetic processes that have analogies with copying errors during textual transmission include recombination (a scribe may switch copying from one manuscript to another), convergence (scribes may independently introduce the same changes that reflect, for example, local dialect) and transposition (!) (when text from one passage is inserted into another). Phylogenetic analyses can be confounded by reversion [166] of a mutation, which is an analogous process to the correction of an error by a copyist.[25]
Based on the occurrence of variants, the next step is to group texts (genomic and literary) with the same variants into families. In both cases, deviation from an ancestral text becomes accentuated with the number of times a text is copied. Biological organisms show increasingly marked genetic divergence as family connectedness becomes more remote: from populations within a species to closely related species (genera), families, orders, and classes of species. In the same way, manuscripts differ from each other and textual critics use metaphors such as family, clan, and tribe to categorise them into related groups.[26]
DNA and textual variants may be classified as progenitor or derived. For example, if a variant X in one or more examples is present only when variant Y is also present, but Y can be present without X, then it appears that Y existed before X, which arose in a text already possessing Y.
A-B-C-D-E-F-G the standard reading in a set of texts
A-B-C-Y-E-F-G population of texts with first mutation Y
A-B-C-Y-E-X-G texts with a second mutation X appearing as a subset in Y
A long-term purpose is the reconstruction of an ancestral text, the progenitor of all the texts which share a set of variants, whether genetic (Figs 2, 4) or literary. The original text giving rise to a family of texts almost certainly no longer exists. In the language of New Testament scholars, the ancestral text might be the “earliest attainable version,”[27] or the vorlage (prototype or template)[28] of a group of texts. Ultimately, it may be possible to reconstruct the ausgangstext (the most recent common ancestor) of them all.[29] The original from the writer’s hand (autograph) [167] may not be reconstructible, but the ultimate aim is to approach it as closely as possible. In the case of biblical textual criticism, “the evidence for the New Testament as a whole is massively strong, and we can be quite sure that, despite lots of small-scale variations here and there, we are reading substantially what the writers intended us to read.”[30]
The ordering of genetic variants enables derivation of a family (phylogenetic) tree (or stemma as a textual critic would say) as exemplified in Figs 1 and 5. An early application of phylogenetic algorithms to literary texts illuminated the history of manuscripts of Chaucer’s Canterbury Tales.[31] The same algorithms have been applied to biblical manuscripts by Jac Perrin and Andrew Edmondson (references cited). An outcome of phylogenetic analysis of old manuscripts of John’s Gospel (“Family 13”) is depicted in Fig. 7. This analysis identifies ten ancient manuscripts in Family 13, which fall into three colour-coded subgroups. The distinctives of Family 13 texts are indicated by comparison with an outgroup (Erasmus’ Textus Receptus, TR, red).
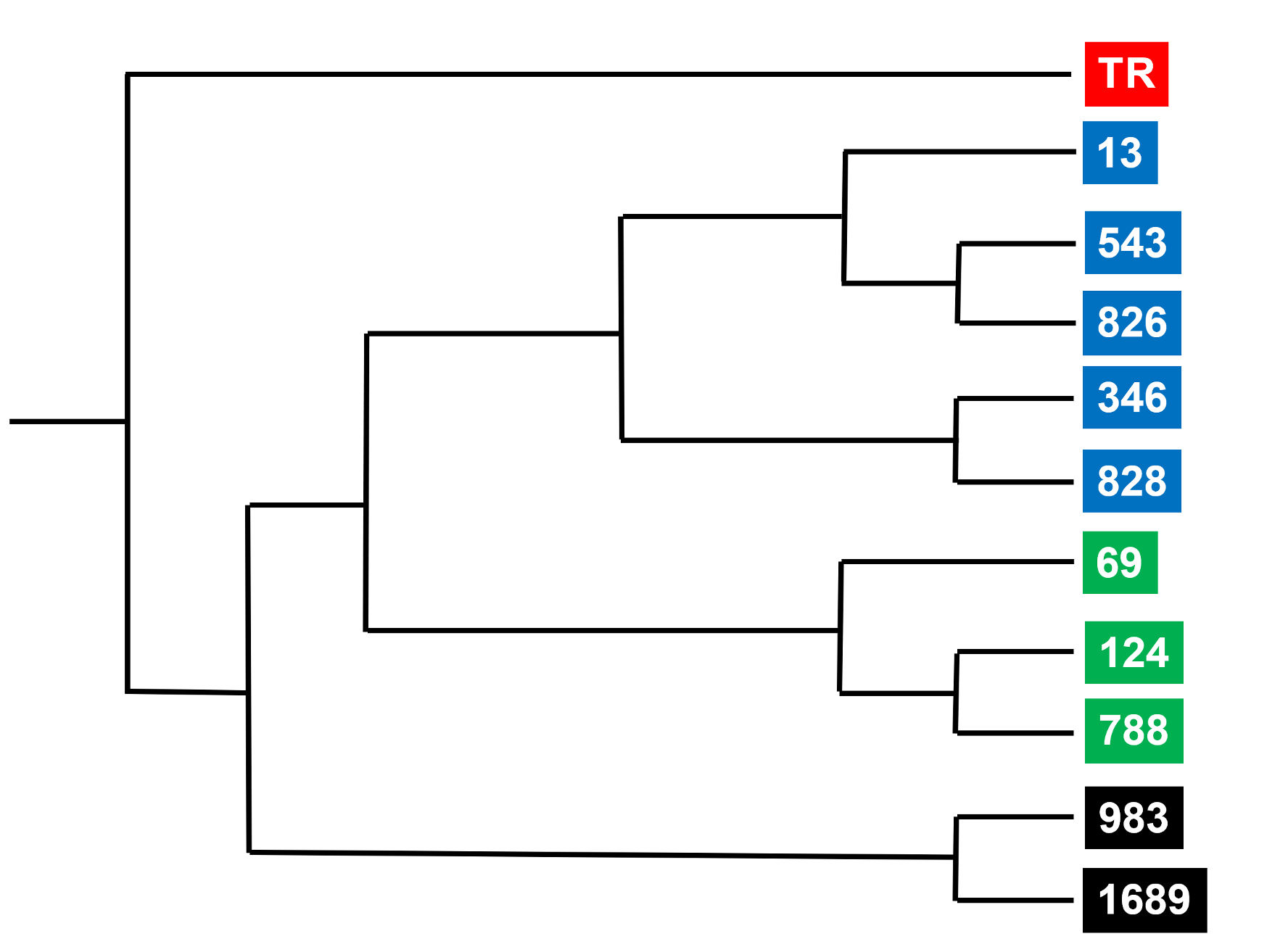
Fig. 7. Relationships of Family 13 manuscripts of John’s Gospel (PAUP* programme) Phylogeny of ten manuscripts of John’s Gospel that belong to Family 13. A standard text (the Textus Receptus, TR; red box) provides a point of comparison, so that characteristics peculiar to Family 13 can be identified. TR provides an outgroup that allows the tree to be rooted at a particular point. From Perrin, “Family 13,” Fig. 62 (https://etheses.bham.ac.uk/id/eprint/4482/), and used with permission of the author.
There are obvious parallels with family trees constructed from karyotype changes or SVA insertions in hominoids. But a simple tree may not provide all the information needed to relate a group of texts. A hybrid manuscript may be produced from several progenitor manuscript traditions. In this case, the SplitsTrees representation is able to depict the mixing of textual material, as in the analysis of Family 13 (Fig. 8). The same subgroups are present, but the network of relationships indicates that the texts were not transmitted in a strictly linear way. Copyists could introduce a variant based on the memory of a manuscript that is different from the one before them. Or they could switch template manuscripts, so that their new manuscript is a hybrid of two or more precursors. We could call this “textual incomplete lineage sorting.” A closer biological analogy might be introgression, the phenomenon by which two emerging species exchange genetic information [168] through intermittent mating.[32] Evolution of species and the development of textual traditions are not strictly linear.
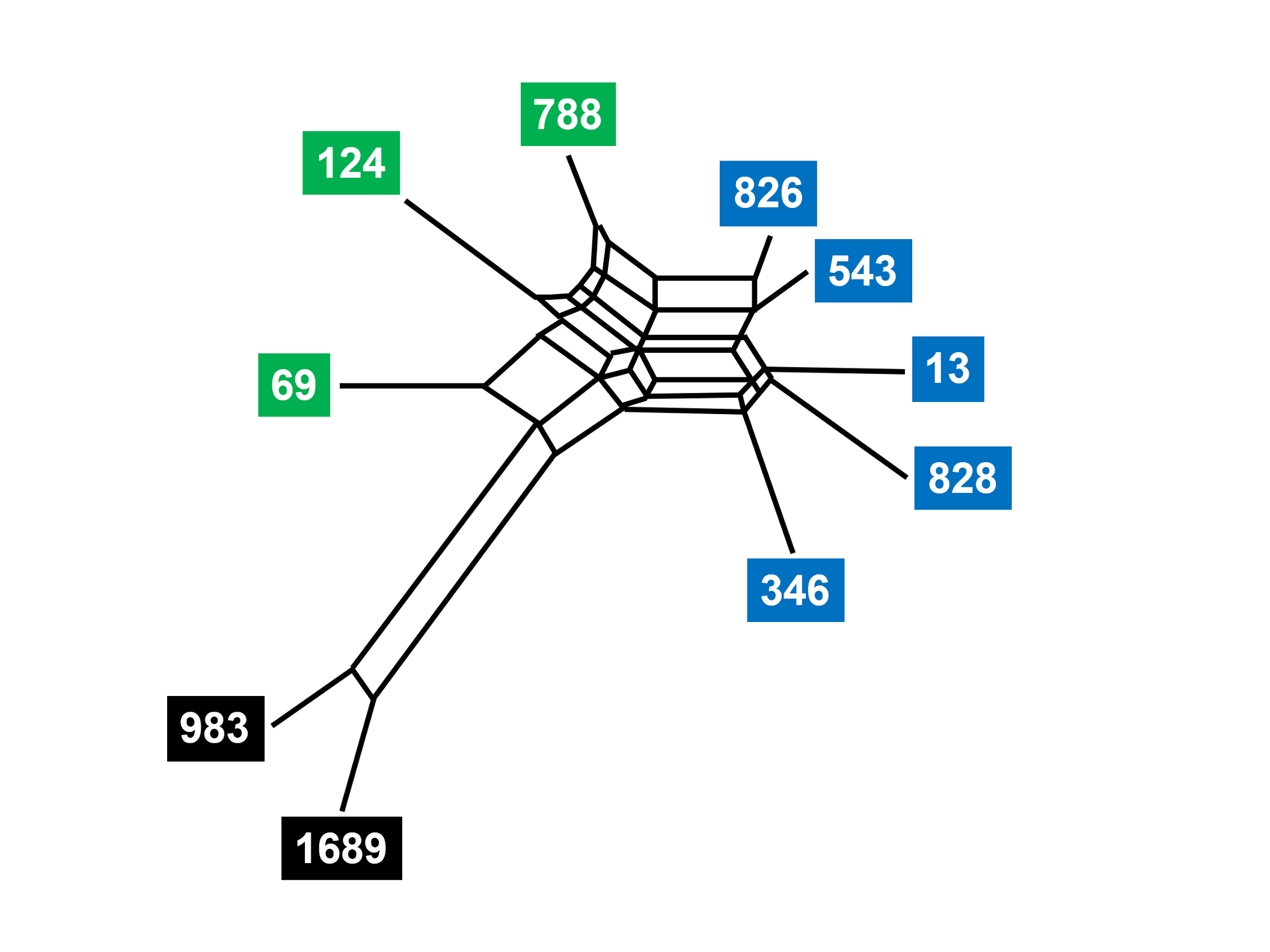
Fig. 8. Relationships of Family 13 manuscripts of John’s Gospel (SplitsTrees programme) Phylogeny of ten manuscripts of John’s Gospel that belong to Family 13. The Textus Receptus provides a point of comparison but is not shown. Modified from Perrin, “Family 13 in Saint John’s Gospel,” Fig. 63 (https://etheses.bham.ac.uk/id/eprint/4482/), and used with permission of the author.
The study of genetic texts is analogous to that of handwritten texts because each presupposes a history. As Perrin has stated, “Ancient manuscripts do not appear ex nihilo”[33]—that is, as if by miracle, or instantaneously by divine fiat. Every artefact is part of a continuum of [169] transmission and can be defined by its evolution and context.[34] In the same way, the myriad genomes (genetic texts) that can be aligned with ours show that they have not appeared ex nihilo. Genomes including ours have developed, by familiar mechanisms, from those of myriad generations of precedents. Genetic analysis is by its nature the deciphering of history.
[170] Genomics reflects textual criticism in another way. Texts can be aligned on the presumption that there is a progenitor urtext from which a family of texts was descended. The fact that we can align genomes thus invites the expectation that there is a genomic urtext from which a group of extant genomes is descended. We could in principle reconstruct a hominoid (ape) urtext belonging to an extinct ape ancestor; or a simian (monkey-ape) urtext or, given a sufficient number of genomes, a primate or mammal or amniote (reptile-bird-mammal) urtext. As described, this reconstruction is well underway with the analysis of karyotypes and of genomes colonised by transposable elements.
Christians need not fear phylogenetic trees. Those constructed from genomes, including our own, are cogent evidence that our genomes are the record of an evolutionary history that is shared with other species. They are but representations of our connectedness with other extant species and point to ancestors that we share with them. Such ancestors are now extinct. They are missing links but are integral to the transformation of one species’ genome into another. But such histories are aspects of God’s creation.
Comparisons with Biblical History
The phylogenetic histories of genomic and literary texts do however differ in one fundamental respect. The former story includes generative trajectories of increasing novelty, complexity, and cognitive capacity. The latter is a degenerative story of progressive alteration of ancestral texts. We must look elsewhere to understand the special nature of genomic history.
Our biological history has analogies with human histories, including those of Israel in the Hebrew Scriptures (or the Christian Old Testament) and of Jesus and his church, as described in the New Testament. There are also differences in these histories. Phylogenetic history has no personal content. There is nothing normative, no moral vision, no intimation of the love or goodness of God, no claim upon our loyalties. And, as read through Christian eyes, Old Testament history [171] points to the history of Jesus—incarnation, mission, resurrection—in which the new creation is inaugurated, and the constitution of reality transformed. “Christ is both the Lord of the whole of the history of created reality and the destiny to which all creation is moving.”[35] In phylogenetics, as in the biblical portrayal of Israel and the church, we are dealing with phases of history.
Biblical faith is irreducibly historical. This character should have given us the a priori expectation that the cosmos and, in our context, biology should also be historical. John Polkinghorne said that it was one of the great discoveries of the twentieth century “that the universe itself has a history and partakes of becoming.”[36] Evolutionary history then presents no challenge to faith in the God who works in history.
All histories are interpreted.[37] The history of Jesus provides the hermeneutic key by which Christians interpret Israel’s history—and the key that enables us to interpret the Primal Testament, evolutionary history as recorded in our DNA, as a part of God’s overarching plan for the world. To St Paul, Christ “is the key that opens all the hidden treasures of wisdom and knowledge.”[38] The history of Jesus brings intelligibility to the histories of the cosmos, of biology, and of Israel. We may see significance, purpose, and hope in the development of our genome that could never be read from the four chemical bases alone.
History is continuous. When the Hebrew Scriptures speak of God’s creation of the cosmos, they often use the participial form, indicating God’s continuing action.[39] This has been called creatio continua.[40] In our spacetime cosmos, time is itself created. From our perspective as players in that cosmos, the continual flow of time—that is, history at any scale or of any entity—is sustained by God. Whether we are looking [172] at the apparently unchanging stars, the development of vertebrate genomes, or a new baby, all are continually given existence by God.
The physical structure of the cosmos is so constituted as to sustain a history (in all its ambiguities) that will lead to the ultimate purposes of God for a redeemed and transformed humanity.
God did what he had purposed and made known to us the secret plan he had already decided to complete by means of Christ. This plan, which God will complete when the time is right, is to bring all creation together, everything in heaven and on earth, with Christ as head.[41]
History is lawful. Both physical and moral cause-and-effect patterns of order are embedded in the universe. To Douglas Spanner, the cosmos is so constituted that its lawful physical processes, as upheld by God, give to nature “a certain built-in autonomy.”[42] Christopher Kaiser has described how the biblical concept of nature’s relative autonomy facilitated the development of science. Nature is self-sufficient because God has granted it laws of operation.[43] God sustains nature in total faithfulness but grants freedom to the creatures (whether atoms, transposable elements, or people) to behave in the ways consistent with their nature.
The relative autonomy of nature reflects the giving of its own order and laws, and the freedom of the cosmos to evolve in accordance with those constraints.[44] The creator sets the parameters, which describe a fruitful universe with the potential to fulfil his purposes of love. At the same time, the creatures are given freedom of action within those limits.
Brueggemann argues that moral law is built into creation. God’s purposes, as given in the Law at Sinai, “are assured in the very fabric [173] of creation.”[45] Keeping the commandments is needed for the viability of creation. God’s commandments “are not social conveniences or conventional rules.” They are “the insistences whereby life in the world is made possible.”[46]
History is contingent. Peter Harrison has stressed the importance of recognising biological evolution as history.[47] Many nineteenth century Christians were perturbed by Darwin’s theory because they thought that the adaptations of organisms demonstrated the elegance of one-off design events. However, if they had only applied God’s action in Israel’s history to God’s action in biological history, they would have seen the same meandering patterns and ambiguous outcomes. Israel’s history was contingent, replete with failure, suffering, and calamity, as well as possessing climaxes of beauty. Our biological history also is contingent, with extinction, disease, and predation as well as much to inspire wonder and praise. The Christian belief that natural history and human history are deeply purposive processes is based on revelation: “what made the case for purpose in history was not a case of logical inferences from available facts, but the revealed tradition contained in the Hebrew Bible and the New Testament.”[48]
Histories show repeating patterns, convergence. In biology, the question has been asked as to what creatures might be generated if the tape of evolution was rerun. Would they be totally different from those that now populate our planet, or would they be similar? Simon Conway Morris has argued that “the number of evolutionary end-points is limited: by no means everything is possible”; and “what is possible has usually been arrived at multiple times, meaning that the emergence of the various biological properties is effectively inevitable.”[49] As three senior physicists state, “Although individual steps in [174] evolution may be random, the overall direction is constrained by the way the world is.”[50]
The same principle seems to hold within the history of human affairs. Free moral choices and visions tend towards certain outcomes. Nick Spencer has suggested that “were we to re-run the tape of Western history, erasing what actually happened and letting it run again, we might, assuming the same deep Christian conditions and commitments, end up with a set of values that, while superficially different, bore a striking resemblance to those we recognize today.”[51]
Some Christians may reject evolution because its process includes random events. But happenstance is inherent to authentic histories. Biological evolution is history, no less than that of manuscript traditions as revealed by textual critics, and in our histories “random seeking leads to non-random finding.”[52] The gospel indicates that there is a destination to world history despite the freedom of its players. The trajectory heading to the purposed climax includes creation’s evolving biota. In Christian terms, therefore, it is reasonable to consider self-aware, worshiping people as the goal of the phylogenetic process. Natural selection has led to our discovery of the personal dimension of reality.[53] We can gladly acknowledge phylogenetic history and its createdness. And the randomness that often characterises our own lives—accident, sickness, struggle—identifies those lives as authentic histories, sustained by their Creator, and destined for transformation in union with Christ.[54]
The author reports there are no competing interests to declare.
Received: 13/09/22 Accepted: 25/11/22 Published: 10/12/22
[1] I thank Dr Liliya Doronina (University of Munster) for permission to use SplitsTree images from her recent publications describing relationships between orders of mammals (refs. 18 and 19), and Dr Roest Crollius (Institut de Biologie de l’École Normale Supérieure) for karyotype images (ref. 11). I am grateful to Rev. Dr Jac Perrin (North Central University, Minnesota) and Dr Andrew Edmondson (University of Birmingham) for permission to use phylogenetic images from their studies on biblical textual criticism.
[2] There is also a tiny chromosome in mitochondria, but this is not relevant to the current discussion.
[3] Roscoe R. Stanyon, Mariano Rocchi, Oronzo Capozzi et al., “Primate Chromosome Evolution: Ancestral Karyotypes, Marker Order and Neocentromeres,” Chromosome Research 16 (2008): 17–39; Steffan Muller, “Primate Chromosome Evolution,” in Genomic Disorders: The Genomic Basis of Disease, ed. James R. Lupski and Pawel Stankiewicz (Totowa: Humana, 2006), 133–152.
[4] Eutherian mammals have a well-developed placenta and include all extant mammals except for monotremes (such as platypus) and marsupials.
[5] Sandra Alverez-Carretero, Asif U. Tamuri, Matteo Battini et al., “A species-level timeline of mammal evolution integrating phylogenomic data,” Nature 602 (2022): 263–267.
[6] Kim Jaebum, Marta Farre, Loretta Auvil et al., “Reconstruction and evolutionary history of eutherian chromosomes,” Proceedings of the National Academy of Sciences of the USA 114 (2017): e5379–88.
[7] Yang Zhou, Linda Shearwin-Whyatt, Jing Li et al., “Platypus and Echidna Genomes Reveal Mammalian Biology and Evolution,” Nature 592 (2021): 756–762.
[8] Darren K. Griffin, Denis M. Larkin, and Rebecca E. O’Conner, “Time Lapse: A Glimpse into Prehistoric Genomics,” European Journal of Medical Genetics 63 (2020): 103640.
[9] More correctly, amphioxus is a sister group to vertebrates. The vertebrate-amphioxus divide has been dated to 684 million years ago.
[10] Paul D. Waters, Hardip R. Patel, Aurora Ruiz-Herrera et al., “Microchromosomes are Building Blocks of Bird, Reptile, and Mammal Chromosomes,” Proceedings of the National Academy of Sciences of the USA 118 (2021): e2112494118.
[11] Nga Thi Thuy Nguyen, Pierre Vincens, Jean Francois Dufayard et al., “Genomicus in 2022: Comparative Tools for Thousands of Genomes and Reconstructed Ancestors,” Nucleic Acids Research 50 (2022): D1025-31; Matthieu Muffato, Alexandra Louis, Nga Thi Thuy Nguyen et al., “Reconstruction of Hundreds of Reference Ancestral Genomes across the Eukaryotic Kingdom,” available at https://www.biorxiv.org/content/10.1101/2022.02.17.480882v1.full.
[12] A detailed presentation of karyotypic evolution in higher primates is provided at the wonderfully interactive and illuminating website https://www.genomicus.bio.ens.psl.eu/genomicus-102.01/cgi-bin/karyotype_handle.pl?numChrom_ref=30&numChrom=30&minGene=50&display=137%3A409%3A413%3A414%3A149%3A150%3A151%3A152%3A&reverse=149&species_id=137
[13] Oleg Simakov, Jessen Bredesen, Kodiac Berkoff et al., “Deeply Conserved Synteny and the Evolution of Metazoan Chromosomes,” Science Advances 8 (2022): eabi5884.
[14] Emma Price, Olympia Gianfrancesco, Patrick T. Harrison et al., “CRISPR Deletion of a SVA Retrotransposon Demonstrates Function as a cis-Regulatory Element at the TRPV1/TRPV3 Intergenic Region,” International Journal of Molecular Sciences 22 (2021): 1911.
[15] Orr Levy, Binyamin A. Knisbacher, Erez Y. Levanon, and Shlomo Havlin, “Integrating Networks and Comparative Genomics Reveals Retroelement Proliferation Dynamics in Hominid Genomes,” Science Advances 3 (2017): e1701256.
[16] In general, over evolutionary timescales, the only stable frequencies of a genetic variant are 0% (the element is lost) or 100% (the element is fixed). This assumes that the element drifts in frequency in a random way.
[17] James K. Schull, Yatish Turakhia, James A. Hemker et al., “Champagne: Automated Whole-Genome Phylogenomic Character Matrix Method Using Large Genomic Indels for Homoplasy-Free Inference,” Genome Biology and Evolution 14 (2022): evac013.
[18] Liliya Doronina, Olga Reising, Hiram Clawson et al., “Euarchontoglires Challenged by Incomplete Lineage Sorting,” Genes 13 (2022): 774.
[19] Liliya Doronina, Graham M. Hughes, Diana Moreno-Santillan et al., “Contradictory Phylogenetic Signals in the Laurasiatheria Anomaly Zone,” Genes 13 (2022): 766.
[20] Any such error occurred once, and its presence in more than one manuscript demonstrates that it was propagated by copying. This is the same logic whereby the presence of “homoplasy-free” mutations (such as ERV and TE inserts) is taken to establish that multiple species have a common ancestor.
[21] Andrew C. Edmondson, “An Analysis of the Coherence-Based Genealogical Method Using Phylogenetics,” PhD Diss. (University of Birmingham, 2018), 166.
[22] Edmondson, “Analysis,” 171–173.
[23] N. T. Wright and Michael Bird, The New Testament in Its World (London: SPCK, 2019), 853.
[24] Yii-Jan Lin, in Edmondson, “Analysis,” 171.
[25] Howe et al., in Edmondson, “Analysis,” 171–172.
[26] Jac D. Perrin Jr, “Family 13 in Saint John’s Gospel,” PhD Diss. (University of Birmingham, 2012), 11.
[27] Perrin, “Family 13,” 10.
[28] Perrin, “Family 13,” 17.
[29] Edmondson, “Analysis,” 13; Wright and Bird, The New Testament, 854.
[30] Wright and Bird, The New Testament in its World, 851.
[31] Adrian C. Barbrook, Christopher J. Howe, Norman Blake, and Peter Robinson, “The phylogeny of The Canterbury Tales,” Nature 394 (1998): 839.
[32] Two populations which are in the process of diverging as new species may undergo backcrossing to form hybrids, with genetic admixture.
[33] Perrin, “Family 13,” 11.
[34] Perrin, “Family 13,” 10.
[35] Adrio Konig, The Eclipse of Christ in Eschatology (Blackwood, South Australia: New Creation, 2007), 31.
[36] John Polkinghorne, Science and Creation (London: SPCK,1988), 39.
[37] N. T. Wright, The New Testament and the People of God (Minneapolis: Fortress, 1992), 88.
[38] Col 2:3.
[39] Walter Brueggemann, Theology of the Old Testament (Minneapolis: Fortress, 1997), 146, 152.
[40] For example, John Polkinghorne, Science and Christian Belief (London: SPCK, 1994), 75–76.
[41] Eph 1:10–11, GNT.
[42] Douglas Spanner, Biblical Creation and the Theory of Evolution (Exeter: Paternoster, 1987), 40.
[43] Christopher Kaiser, Creation and the History of Science (London: Marshal Pickering, 1991), 15–34.
[44] As propounded no later than John Philoponus in the sixth century. See Harold Turner, The Roots of Science (Auckland: DeepSight Trust, 1998), 101.
[45] Brueggemann, Theology, 303.
[46] Brueggemann, Theology, 201.
[47] Peter Harrison, “Evolution, Providence and the Problem of Chance,” in Abraham’s Dice, ed. Karl W. Giberson (Oxford University Press, 2016), 260–290.
[48] Harrison, “Evolution,” 279.
[49] Simon Conway Morris, Life’s Solution: Inevitable Humans in a Lonely Universe (Cambridge University Press, 2003), xii–xiii.
[50] Andrew Briggs, Hans Halvorson, and Andrew Steane, It Keeps Me Seeking (Oxford University Press, 2018), 63.
[51] Nick Spencer, The Evolution of the West (Louisville: Westminster John Knox, 2018), 8.
[52] Andrew Steane, Faithful to Science (Oxford University Press, 2014), 71.
[53] Briggs et al., It Keeps Me Seeking, 188.
[54] 1 Cor 2:7–9; Rom 5:3–5; 8:35–39; Jas 1:2–4, 12.